Registration of both local and foreign-manufactured medical devices in Malaysia is generally a two-part process. They are mainly regulated by the Medical Device Authority (MDA) of Malaysia, a local regulation and authorization body. However, medium to higher risk medical devices, falling under risk classification of Class B, C or D, are required to go through an additional safety and performance conformity assessment by an MDA-certified third-party in Malaysia, before they can be submitted to MDA for product registration application.
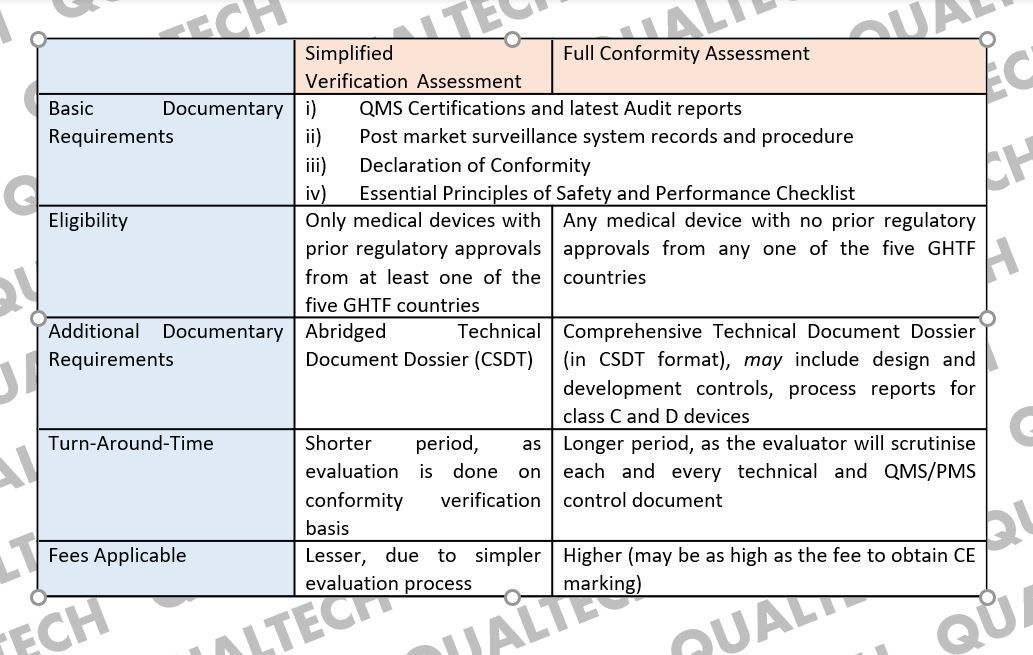
Conformity Assessment Bodies (CAB), which are the MDA-certified third-party in Malaysia, provide two different medical device conformity assessment services, i.e. simplified verification (SV) assessment and full conformity (FC) assessment. Local and foreign manufacturers who have obtained prior medical device registration approval from at least one of the five GHTF countries, i.e. European Union, USA, Japan, Australia and Canada, are eligible for the much simpler pathway of SV assessment while the others must go through the extensive FC assessment, where the documentary requirements are similar to obtaining CE certification.
Conformity Assessment Process in Malaysia
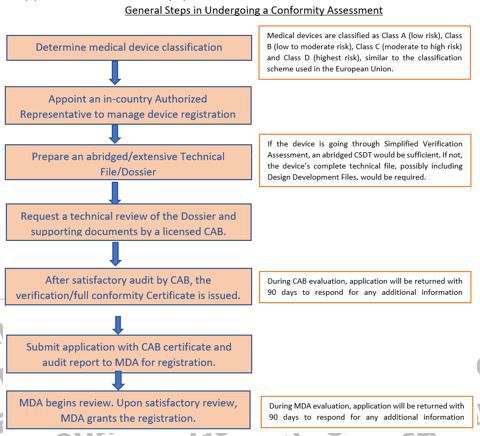
Conformity assessment is a systematic and ongoing examination of evidence and procedures to ensure the safety, performance, benefit and risk of medical devices. It is also to ensure manufacturing compliance to essential principles of safety and performance (EPSP) and requirements of the Medical Device Act 2012 (Act 737). The classification of a medical device determines the conformity assessment procedures to be undertaken. Conformity assessment becomes more stringent as the risk of medical device increases.
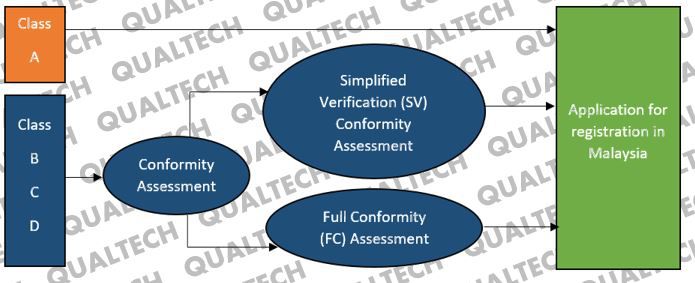
All class B, C, and D medical devices must first undergo conformity assessment before they can be submitted for evaluation with the MDA. Owing to low risk potential, class A medical devices have been exempted from being conformity-assessed by a CAB prior to submission with the MDA.
The involvement of a third party in the registration process is uncommon in other countries where the application for device registration is submitted to the authority alone to be processed for conformity assessment. Recently India’s medical device regulatory body, CDSCOm came up with regulation for all medical devices to be assessed by locally-registered Notified Bodies before submitting product registering applications with the CDSCO. Thus, the CAB’s involvement in the Malaysian medical device registration process has lengthened the time required for medical device approval. Due to the high demand by applicants, conformity assessment takes on average two months, sometimes longer, depending on the risk classification. This may be an important consideration for product registration planning.
Conformity Assessment Bodies are selected and licensed by the MDA to provide two types of assessment services for medical devices, i.e. simplified verification, or full conformity assessment, before the device can be submitted for registration. Currently there are 17 approved and licensed CABs for applicants to choose from and each of them has a specific scope or expertise in particular technical areas in Malaysia. There are the locally-established ones such as Sirim QAS International Sdn. Bhd., Genuine Diamond Sdn. Bhd. and Medcert Malaysia Sdn. Bhd. and then the local-subsidiaries of international Notified Bodies such as Tüv Süd (Malaysia) Sdn. Bhd., BSI Services Malaysia Sdn. Bhd and SGS Malaysia Sdn. Bhd. Therefore, it is important that applicants confirm that their chosen CAB covers the scope of their device before selecting them. Recently, MDA released a Guidance Document on the ‘Conformity Assessment of Medical Devices’, numbered as MDA/GD/0031. This document provides guidance to applicants and CAB to understand the assessment elements that are tabulated clearly in the document.
Types of CAB assessments – Simplified Verification & Full Conformity
Essentially, all conformity assessments, be it following the SV assessment or FC assessment route, comprise of assessing the following elements of a medical device, for the purpose of medical device registration.
(a) Quality management system (QMS) of the manufacturer (and any sub-contractors);
(b) A system for post-market surveillance of the device;
(c) Technical documentation (also-called as Common Submission Dossier Template (CSDT) in Malaysia); and
(d) A declaration of conformity by the manufacturer.
General Steps in Undergoing a Conformity Assessment
✦ YOUR DEVICE IS ELIGIBLE TO GO THROUGH SIMPLIFIED VERIFICATION ASSESSMENT ✦
Simplified verification assessment is much simpler and conducted over a shorter period of time, than full conformity assessment. All class B, C and D medical devices can qualify for simplified verification assessment, provided they adhere to the requirements set by MDA.
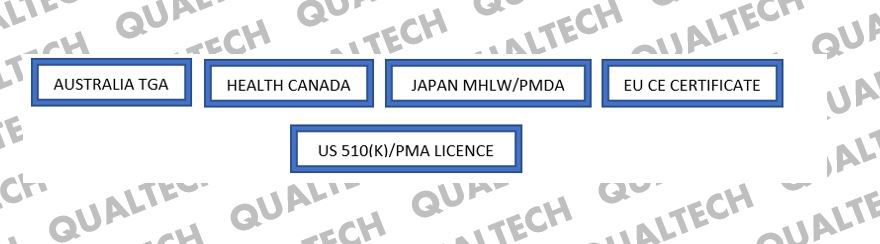
Any medical device which has had prior valid registration approval(s) in at least one of the five Global Harmonization Task Force (GHTF) founder countries can qualify to undergo simplified verification assessment by a CAB, prior to submission with MDA. These five countries are Australia, Canada, Japan, the European Union, and the United States of America. MDA recognises medical device registration approvals granted by the medical device regulating bodies of these 5-member countries, and as such paves a way for these medical devices to be registered much faster and simpler, compared to devices with no prior approvals from any one of these countries. These devices’ CSDT will only be verified for its conformity with international and local standards they claim to abide in the Essential Principles Requirement document, along with the manufacturer’s establishment of a valid quality management system and post market surveillance system. Simplified verification assessment allows the applicant to submit a concise CSDT for verification-purposes, as opposed to full conformity assessment which will require a more elaborate set of documents.
* Full conformity assessment
Full conformity (FC) assessment, on the other hand, are reserved for medical devices with no prior registration approval in any of the 5-GHTF founder countries. To say it simple, the documentary requirement for a class B, C or D medical device to undergo FC assessment is highly similar to the process of obtaining CE certification, including detailed post market complaints/FCA records. It is also a more stringent process as the CAB will request for each and every technical document, as per each local and international standards the device complies with. Even if they are not readily available in the English language, CAB requires all of them to be translated for the evaluation process, which is both time and cost-consuming.
Another point to note is, even if it has been registered and marketed in any other countries globally, as long as the simplified verification assessment’s requirements are not fulfilled, FC assessment is the only option available. To verify a device’s conformity to the relevant standards it is expected to comply, CAB will conduct extensive audits for all the elements to be assessed, as set by MDA. If there are any non-conformities found in the QMS, PMS system and complete device dossier (including each and every test report), there is a high possibility for the application to be rejected by the CAB, non-refundable.
Full conformity assessment in Malaysia is similar to CE certification or US FDA premarket approval application. No predicate device or prior approval can be used as a leverage for this approval or to follow a simplified registration process. FC assessment conducts a thorough scientific and regulatory review to evaluate the safety and effectiveness of medical devices, regardless of risk classification.
a) Quality management system (QMS): CAB will consider any relevant existing certification and, if not satisfied, may carry out an on-site audit of the manufacturer’s facility. Criteria for acceptance of existing certification shall include the following;
a. QMS certificate issued by MDA registered CAB;
b. QMS certificate issued by any notified body listed in New Approach Notified and Designated (NANDO);
c. QMS certificate issued by certification bodies from reference regulatory countries (US, Canada, Australia, Japan, EU);
b) Post-market surveillance system (PMS): CAB will ensure PMS is established, maintained and implemented by the establishment. The following processes shall be documented, maintained and implemented by the establishment:
a. complaint handling;
b. distribution records;
c. mandatory problem/adverse event reporting;
d. field corrective action; and
e. recall.
c) Technical documentation: A complete CSDT must be established by the manufacturer. No abridged version will be accepted. Each and every test report and clinical evaluation reports must be made available and submitted when requested, for the CAB to scrutinise and evaluate thoroughly. CAB determines the adequacy of the documented evidence in support of the manufacturer’s declaration of conformity to the EPSP through a review of the CSDT and technical documentation.
d) Declaration of conformity: Medical Devices manufacturers shall attest that its medical device complies fully with all applicable Essential Principles for Safety and Performance and other requirements of Act 737 and the subsidiary regulations under it, documented in a written ‘Declaration of Conformity’ (DOC).
Furthermore, even though the device’s evaluation process is similar to obtaining CE marking, full conformity assessment certification obtained in Malaysia cannot be used with any other regulatory agencies as a reference approval. This is a huge drawback, as the turn-around-time and fees applicable are similar this certification to obtain CE marking and the latter can be used as refence agency approval in many other ASEAN countries. It is highly recommended for a new medical device to go for CE marking approval first as it can expedite the product registration process in various ASEAN countries.
Turn-around-time for conformity assessments with CAB
There is no specific turn-around time set by MDA for CABs to follow, for both simplified verification assessment and full conformity assessment. Each CAB has their own timeline, which they’d follow, according to the time required by the lead auditor to conduct the assessment. Generally, if the abridged dossier and relevant supporting documents are all complete, simplified verification assessment would take approximately 3 months to be completed, sometimes shorter or longer. As for FC assessment, the audit periods depend on the risk classification of the device evaluated and comprehensiveness of the CSDT submitted, not taking into consideration any stop-clock-time in between.
Fees applicable for conformity assessments with CAB
The fees applicable for a medical device to undergo simplified verification assessment or full conformity assessment hasn’t been set by MDA, so just like turn-around-time, each CAB has their own quotation to follow. Generally, fees to conduct FCA in Malaysia is similar to obtain the device’s CE marking/certification, depending on the risk classification.
So, to summarise the similarities and differences between Simplified Verification Assessment and Full Conformity Assessment:
With a local office in Kuala Lumpur, Qualtech can assist with your registration and interact with the Medical Device Authority on your behalf.
Our team of expert consultants in Qualtech Malaysia are fully capable of acting as your Malaysia Authorized Representative if you have no local office in the country or simply wish to outsource the regulatory functions of a local office. We can also help you access other medical device markets in Southeast Asia according to your business strategy.
Malaysia is a one of the most robust and growing medical markets in ASEAN. Let Qualtech help you access this growing market.